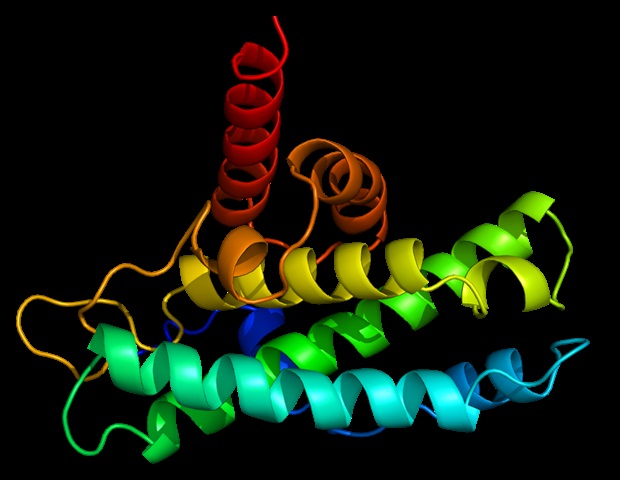
A study from Bochum describes a mammal-specific domain of the prion protein and offers new approaches for research into neurodegenerative diseases. At first, they cause memory deficits and difficulties in walking, finally they inhibit elementary motor skills and destroy basic brain functions: Prion diseases are progressive and invariably fatal neurodegenerative diseases. They are caused by misfolded prion proteins.
The mechanisms behind their development are still poorly understood. Researchers at Ruhr University Bochum, Germany, have now discovered a new region in prion proteins and thus identified a potential new area for research into neurodegenerative diseases. This domain is specific to mammalian prion proteins and influences the aggregation of the protein.

The neuroscientists published their research results on April 22, 2024 in the Journal of Biological Chemistry. Prion diseases are very rare. About two in a million people are affected.
One of the best-known examples is Creutzfeldt-Jakob disease, which gained particular prominence during the BSE crisis in the 1990s. As the disease progresses, the brain takes on a sponge-like perforated structure and shows protein deposits. "The disease is triggered by the misfolding of the cellular prion protein, PrP for short, in the brain," explains Professor Jörg Tatzelt from the Institute of Biochemistry and Pathobiochemistry at Ruhr University Bochum, who led the study.
"The pathological proteins accumulate in the brain and form.